 |
|
|
|
|
|
|
Thalassemia Minor Thalassemia Minor
Thalassemia Major is present at birth, and the child cannot outgrow it. Thalassemia cannot be 'contracted'. It is not infectious or contagious. Both parents need to have Thalassemia trait in order for the child to contract it. If a person with a Thalassemia trait marries another such person, the offspring has a 25% chance of developing full blown Thalassemia. If only one parent has the trait, the trait may be passed on to the child. A person with a Thalassemia trait shows no symptoms except perhaps for anaemia. Iron supplements don't help, and should not be taken. Having a Thalassemia trait does not make a person susceptible to any diseases. This trait does not harm the person in any way, and the person can live a perfectly normal life without even being aware that he has a Thalassemia trait. A Thalassemia trait will NOT develop into the full blown disease at any stage in life. Thalassemia Minor |
|
There are two forms of beta thalassemia. They are thalassemia minor and thalassemia major (which is also called Cooley's anemia).
Thalassemia minor: The individual with thalassemia minor has only one copy of the beta thalassemia gene (together with one perfectly normal beta-chain gene). The person is said to be heterozygous for beta thalassemia.
Persons with thalassemia minor have (at most) mild anemia (with slight lowering of the hemoglobin level in the blood). This situation can very closely resemble that with mild iron-deficiency anemia. However, persons with thalassemia minor have a normal blood iron level (unless they have are iron deficient for other reasons). No treatment is necessary for thalassemia minor. In particular, iron is neither necessary nor advised.
Thalassemia Minor often coexists
with other diseases such as asthma, and mood disorders
Thalassemia minor is nothing
to do with conception. It should be looked seperately for different causes.
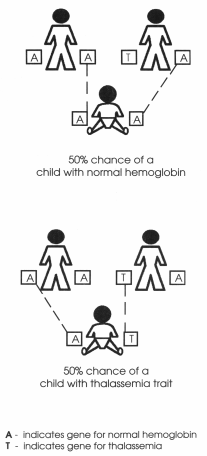
the only thing to know is t6hat if both husband and wife are Thalassemia
carrier then in each pregnancy there is 25% chance of having a thalassemia
major child, 50% chance of having Thalassemia minor/carrier child and 25%
"Normal-not even a carrier"
Dr J.S. Arora
General Secretary
National Thalassemia
Welfare Society &
Federation of Indian
Thalassemics
|
|
|
|
|
| Thalassemia Minor is
the mildest form of the illness. The individuals may be heterozygous and
inheriting only one mutant or p-thal gene, which limits the synthesis of
beta-peptide chains. Or they may have inherited a genetically distinct
abnormality of p-globin chain which produces a milder disease. The gene
for alpha chain synthesis is normal. There is usually compensatory increase
in delta chain synthesis, leading to levels of Hb A2 being higher than
normal. Hemoglobin A2 may be markedly reduced with co-existing iron-deficiency
anemia.
Increase in the level of hemoglobin F is inconstant. Production of hemoglobin A is only slightly reduced. Thalassemia Minor patients have only a very mild anemia, sometimes with abdominal pain and mild icteric, tinge. Thalassemia minor is often confused with iron deficiency anemia and is treated as such, till the correct diagnosis is suspected. Serum iron concentration is on the higher side and iron binding capacity is reduced. Free erythrocyte porphyrin is normal in thalassemia minor (30 microgram/dL of whole blood). This is moderately raised in iron deficiency anemia (30 to 190 ug/ dL of whole blood). This is a useful diagnostic test to differentiate iron deficiency states from thalassemia minor and lead poisoning. ALPHA-THALASSEMIA-HEMOGLOBIN H DISEASE The synthesis of alpha-peptide chains is suppressed. Defective alpha chain synthesis effects the production of all the normal hemoglobin viz., A, A2 and F. Four beta-peptide chains polymerise to a tetrameric form giving rise to hemoglobin H. Heterozygous alpha thalassemia is usually very mild and is often not associated with any regular alteration in the hemoglobin pattern in the adult life. There are possibly two alleles for alpha-thalassemia gene viz., alpha-thalassemia1 causing complete inhibition of alpha chain synthesis and alpha-thai2 causing only impaired synthesis of alpha chains. Alpha-thalassemia is most prevalent in the countries of South East Asia. The infant is usually normal at birth. The mainstay of managing
these cases is repeated blood transfusions. An attempt should be made to
maintain hemoglobin level above 10-12 g/dL (by hyper transfusion) to ensure
active life and adequate growth. Group and type specific, fresh, triple
saline washed, packed red cell transfusions are the most desired form of
component therapy. These are to be transfused at the rate of 10-15 ml/kg
every 2 to 3 weeks. Blood transfusions may result in hemolytic or febrile
reactions, transmission of viral infections (HIV1 & 2, Hepatitis B
and C, Cytomegalovirus) and iron overload. Routine donor screening for
these viral infections is a must. All thalassemics should be vaccinated
with Hepatitis B vaccine before starting transfusions. Iron overload results
in multiple organ dysfunction due to hemosiderosis and hemochromatosis.
Chronic anemia itself is a potent stimulator of iron absorption from the
gut. Iron overload can be reduced by regular chelation therapy and reducing
iron absorption by keeping the hemoglobin levels high.
At present only desferrioxamine
is available as iron chelating agent in parenteral form. It should be given
as continuous subcutaneous infusion in the dose of 25-50 mg/kg/day over
a period of 8-12 hours, during the night, by specially designed microinfusion
pumps, (at least 5-6 nights per week). The chelation should start by 12-14th
transfusion. 100 mg of vitamin C daily should be concurrently administered.
Overdose of desferrioxamine results in growth retardation, visual and auditory
toxicity. Cataracts have been noted in some children on long term desferrioxamine
therapy. Deferiprone (DFP) is an effective oral iron chelating agent with
minimal toxicity. It is given in a dose of 75 mg/kg/day in 2-4 divided
doses. The most common side-effect is arthropathy. Other oral chelators
include pyridoxine hydrazine, HBED and desferrothiocine. These are still
under trials.
|
|
||||
 |
| Privacy Policy for LiveIndia.Com |

Multi Dimensional Entertainment magazine from INDIA |
 |









